---
title: "genemodel Tutorial"
author: "J Grey Monroe"
date: "`r Sys.Date()`"
output: rmarkdown::html_vignette
vignette: >
%\VignetteIndexEntry{genemodel Tutorial}
%\VignetteEngine{knitr::rmarkdown}
%\VignetteEncoding{UTF-8}
---
```{r, echo=F}
library(genemodel)
```
This package provides a simple way to make positionally accurate plots of gene models similar to those found on The Arabidopsis Information Resource (TAIR). These plots are suitable for presentations and publications where showing gene models is appropriate to comunicate genetic research.
### Installation
To install the package:
```{r, eval=F}
install_bitbucket("greymonroe/genemodel")
library(genemodel)
```
### Example
Now lets look at an example gene that we are going to model. This is gene is `AT5G62640`. The original gene model information can be found here http://www.arabidopsis.org/servlets/TairObject?type=gene&id=1000654517. It is stored in the `Gene Feature` table on that page.
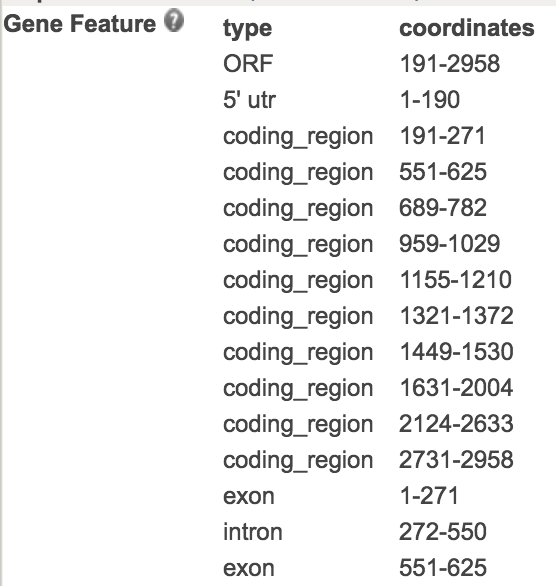 ...
Once this table is extracted and saved as .csv or .txt table file, it can be loaded into R as a data.frame object. The package *genemodel* has this data already stored and can be loaded like this:
```{r, eval=T}
data("AT5G62640")
```
When we look at the strucutre of this data.frame,
```{r, eval=T}
head(AT5G62640, 15)
```
we see that it is a two column data.frame with the first column, "*type*," containing the name of the feature type and the second,"*coordinates*," specifying the coordinate position within the gene model that the feature occupies.
The feature types that TAIR provides in the `Gene Feature` table for gene models are:
* ORF - the Open Reading Frame which extends from the first to last exon
* Coding Region - segments of the gene that code RNA
* 5` UTR - Untranlated Region.
* Exon
* Intron
* 3` UTR
### genemodel.plot function
Before we can plot the gene, we need to extract some other information from the TAIR gene model description. First we need the start and stop base pair positions for the gene. These can be found in the `Map Locations` section of the TAIR gene model page and correspond to the `Coordinates` of `Map Type` 'nuc_sequence.' Next weed the direction of transcription which can also be found in the `Map Locations` section under `Orientation`. Again we want the value for corresponding the `Map Type` that equals 'nuc_sequence' See image below from TAIR.
...
Once this table is extracted and saved as .csv or .txt table file, it can be loaded into R as a data.frame object. The package *genemodel* has this data already stored and can be loaded like this:
```{r, eval=T}
data("AT5G62640")
```
When we look at the strucutre of this data.frame,
```{r, eval=T}
head(AT5G62640, 15)
```
we see that it is a two column data.frame with the first column, "*type*," containing the name of the feature type and the second,"*coordinates*," specifying the coordinate position within the gene model that the feature occupies.
The feature types that TAIR provides in the `Gene Feature` table for gene models are:
* ORF - the Open Reading Frame which extends from the first to last exon
* Coding Region - segments of the gene that code RNA
* 5` UTR - Untranlated Region.
* Exon
* Intron
* 3` UTR
### genemodel.plot function
Before we can plot the gene, we need to extract some other information from the TAIR gene model description. First we need the start and stop base pair positions for the gene. These can be found in the `Map Locations` section of the TAIR gene model page and correspond to the `Coordinates` of `Map Type` 'nuc_sequence.' Next weed the direction of transcription which can also be found in the `Map Locations` section under `Orientation`. Again we want the value for corresponding the `Map Type` that equals 'nuc_sequence' See image below from TAIR.
 We now have the information necessary to plot the gene with `genemodel.plot`
```{r, fig.width=7.5, fig.height=2}
genemodel.plot(model=AT5G62640, start=25149433, bpstop=25152541, orientation="reverse", xaxis=T)
```
`genemodel.plot` automatically recongizes the types of gene features found in TAIR gene models and plots them in accurate positions and orientation. By default, UTRs are colored light blue, exons are colored dark blue and introns are indicated by the bent line. The direction of transcripton is also marked in way consitent with TAIR notation by the pointed end of the gene model. By only plotting UTRs, coding region and introns, `genemodel.plot` ignores the 'ORF' and "exon' feature types as they are redundant.
### Alternative splicing
With a little creativity, it is easy to imagine using genemodel to plot such things as alternative splicing. For example, an exon and it's neighboring introns can be removed and replaced by a sinlge intron to create a plot showing a different splice variant.
```{r}
spl1<-data.frame(
type=c("5' utr", "coding_region", "intron", "coding_region", "intron", "coding_region","3' utr"),
coordinates=c("1-50", "50-100", "100-150", "150-200", "200-250", "250-300","300-350"))
spl2<-data.frame(
type=c("5' utr", "coding_region", "intron","coding_region","3' utr"), coordinates=c("1-50", "50-100", "100-250", "250-300","300-350"))
par(mfrow=c(2,1))
genemodel.plot(model=spl1, start=1, bpstop=350, orientation="reverse", xaxis=T)
genemodel.plot(model=spl2, start=1, bpstop=350, orientation="reverse", xaxis=F)
```
## mutation.plot function
The next function we will look at is the `mutation.plot` function which plots mutations at correct positions on an already plotted gene model.
```{r, fig.width=7.5, fig.height=2}
genemodel.plot(model=AT5G62640, start=25149433, bpstop=25152541, orientation="reverse", xaxis=T)
mutation.plot(25150214, 25150214, text="P->S", col="black", drop=-.15, haplotypes=c("red", "blue"))
mutation.plot(25150659, 25150659, text="V->S", col="black", drop=-.15, haplotypes=c("red"))
mutation.plot(25150639, 25150639, text="L->P", col="black", drop=-.35, haplotypes=c("blue"))
```
`mutation.plot` adds mutations to a prexisting gene model plot. In this example, amino acid substitutions are shown at exact positions. The colored dots correspond to the hapolotype group that has this mutation. The `drop` parameter can be used to offset the positionin of close mutation for easy visualization.
We now have the information necessary to plot the gene with `genemodel.plot`
```{r, fig.width=7.5, fig.height=2}
genemodel.plot(model=AT5G62640, start=25149433, bpstop=25152541, orientation="reverse", xaxis=T)
```
`genemodel.plot` automatically recongizes the types of gene features found in TAIR gene models and plots them in accurate positions and orientation. By default, UTRs are colored light blue, exons are colored dark blue and introns are indicated by the bent line. The direction of transcripton is also marked in way consitent with TAIR notation by the pointed end of the gene model. By only plotting UTRs, coding region and introns, `genemodel.plot` ignores the 'ORF' and "exon' feature types as they are redundant.
### Alternative splicing
With a little creativity, it is easy to imagine using genemodel to plot such things as alternative splicing. For example, an exon and it's neighboring introns can be removed and replaced by a sinlge intron to create a plot showing a different splice variant.
```{r}
spl1<-data.frame(
type=c("5' utr", "coding_region", "intron", "coding_region", "intron", "coding_region","3' utr"),
coordinates=c("1-50", "50-100", "100-150", "150-200", "200-250", "250-300","300-350"))
spl2<-data.frame(
type=c("5' utr", "coding_region", "intron","coding_region","3' utr"), coordinates=c("1-50", "50-100", "100-250", "250-300","300-350"))
par(mfrow=c(2,1))
genemodel.plot(model=spl1, start=1, bpstop=350, orientation="reverse", xaxis=T)
genemodel.plot(model=spl2, start=1, bpstop=350, orientation="reverse", xaxis=F)
```
## mutation.plot function
The next function we will look at is the `mutation.plot` function which plots mutations at correct positions on an already plotted gene model.
```{r, fig.width=7.5, fig.height=2}
genemodel.plot(model=AT5G62640, start=25149433, bpstop=25152541, orientation="reverse", xaxis=T)
mutation.plot(25150214, 25150214, text="P->S", col="black", drop=-.15, haplotypes=c("red", "blue"))
mutation.plot(25150659, 25150659, text="V->S", col="black", drop=-.15, haplotypes=c("red"))
mutation.plot(25150639, 25150639, text="L->P", col="black", drop=-.35, haplotypes=c("blue"))
```
`mutation.plot` adds mutations to a prexisting gene model plot. In this example, amino acid substitutions are shown at exact positions. The colored dots correspond to the hapolotype group that has this mutation. The `drop` parameter can be used to offset the positionin of close mutation for easy visualization.